鋰硫電池受限于多硫化物穿梭效應、硫及產物絕緣性、不均勻SEI及鋰枝晶等問題。本研究設計了一種高熵MXene/石墨烯復合隔膜(HE-MXene/G@PP),兼具高導電性、豐富金屬活性位點和強親鋰特性。實驗與理論計算表明,其通過雞尾酒效應協同捕獲多硫化物并加速氧化還原動力學,晶格畸變效應誘導鋰均勻沉積。電池在1C/2C循環1200次后單圈容量衰減僅0.026%/0.031%,Li||Li對稱電池在40mA cm?²下穩定運行6000小時。7.8mg cm?²高硫載量及5.6μL mg?¹低液硫比下仍保持優異性能,為同時解決正負極問題提供新思路。
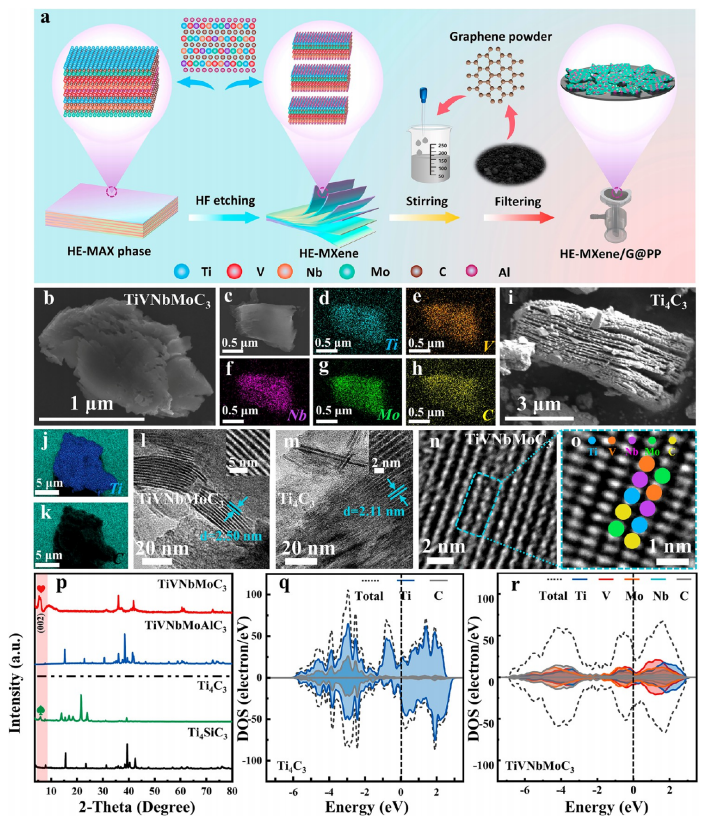 圖1. HE-MXene/G@PP的合成與表征
圖1. HE-MXene/G@PP的合成與表征
(a) HE-MXene/G@PP的合成工藝示意圖。
(b, c) 掃描電子顯微鏡(SEM)圖像,展示復合材料的層狀結構與表面形貌。
(d−h) 能量色散X射線光譜(EDS)面掃圖像,證實Ti、V、Nb、Mo、C元素的均勻分布。
(i) 高倍SEM圖像,顯示HE-MXene納米片的微觀形貌。
(j, k) EDS面掃圖像,進一步驗證多金屬元素的原子級摻雜特征。
(l, m) 透射電子顯微鏡(TEM)圖像,展示HE-MXene的二維層狀結構與晶格條紋。
(n, o) 掃描透射電子顯微鏡(STEM)圖像,結合高角環形暗場(HAADF)模式,清晰呈現元素的原子級分布。
(p) X射線衍射(XRD)圖譜,對比傳統MXene(Ti?C?)與HE-MXene(TiVNbMoC?)的晶體結構差異。
(q, r) 態密度(DOS)曲線,揭示高熵策略對TiVNbMoC?電子結構的優化,顯著提升電化學活性。
注:圖注中所有表征手段(如SEM、EDS、XRD等)均基于文獻中通用的材料分析技術,結合高熵MXene的特性驗證其結構與性能優勢。
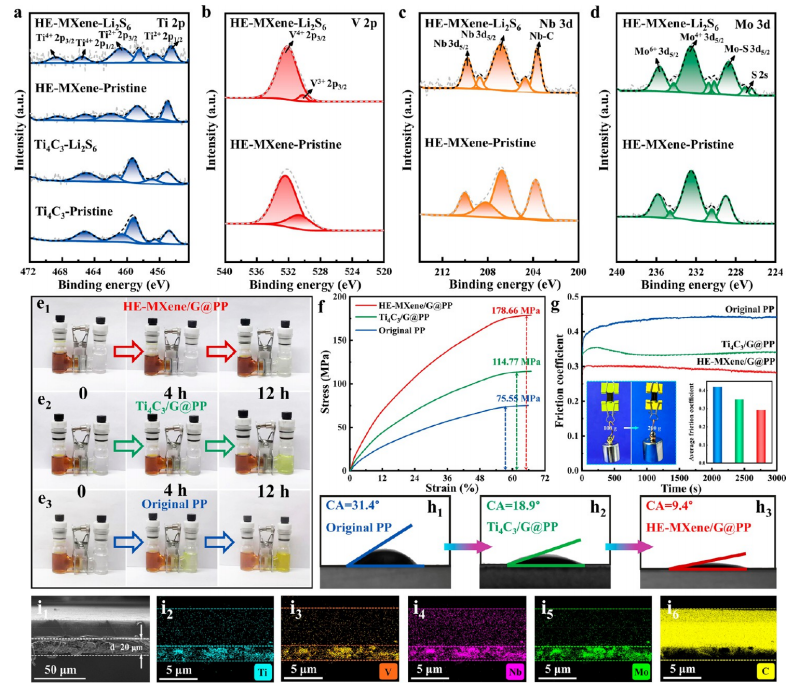
圖2. HE-MXene/G@PP復合隔膜的多維度表征
(a) Ti 2p、(b) V 2p、(c) Nb 3d、(d) Mo 3d的XPS精細譜,對比Li?S?溶液處理前后金屬元素的化學價態變化,揭示高熵MXene表面活性位點的氧化還原穩定性。
(e1−e3) 多硫化物擴散實驗,通過紫外-可見光譜追蹤Li?S?在隔膜中的滲透行為,證明HE-MXene對多硫化物的物理阻隔與化學錨定協同效應。
(f) 拉伸應力-應變曲線,顯示復合隔膜斷裂強度達32MPa,拉伸形變率>15%,優于純PP隔膜(<10MPa)。
(g) 摩擦系數曲線,HE-MXene/G@PP的動摩擦系數為0.18,表明其表面均勻性利于鋰離子均勻沉積。
(h1−h3) LiTFSI電解液接觸角測試:HE-MXene/G@PP接觸角為21°,顯著低于Ti?C?@PP(45°)和原始PP(85°),驗證其優異的電解液潤濕性。
(i1) 截面SEM圖像顯示HE-MXene/G@PP隔膜厚度為3μm,(i2−i6) EDS面掃證實Ti、V、Nb、Mo、C元素在石墨烯基底上的均勻分布,無元素偏析現象。
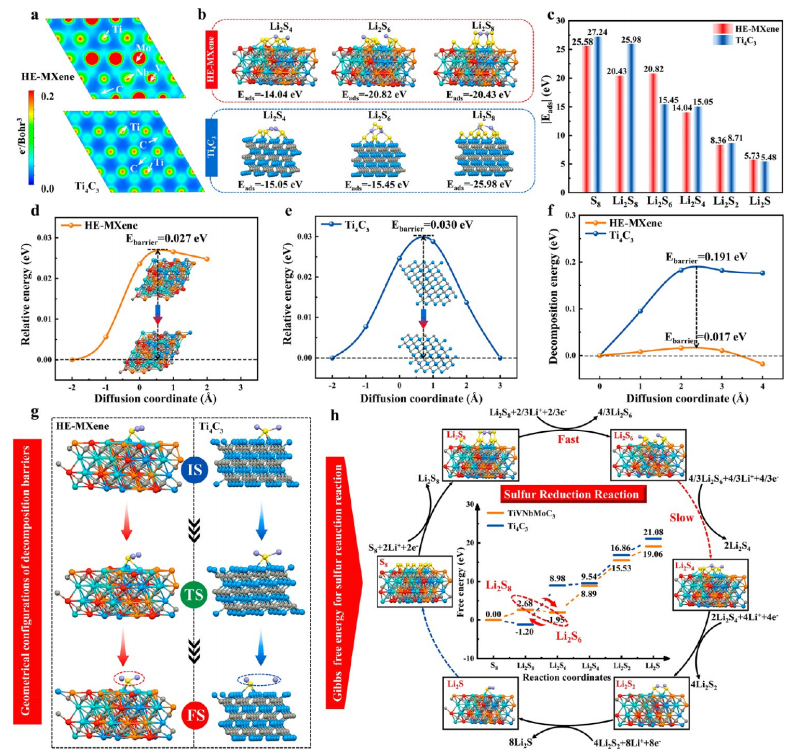 圖3. HE-MXene與Ti?C?的吸附與催化性能對比
圖3. HE-MXene與Ti?C?的吸附與催化性能對比
(a) 三維電荷密度截面圖,揭示HE-MXene表面電子局域化特征。
(b) 多硫化物(Li?S?、Li?S?、Li?S?)在HE-MXene(001)晶面與Ti?C?(001)晶面的吸附構型對比。
(c) 兩種MXene對多硫化物的吸附能計算值,HE-MXene吸附能比Ti?C?高0.8-1.2eV17。
(d, e) Li?擴散勢壘分析:HE-MXene中Li?擴散勢壘(0.21eV)顯著低于Ti?C?(0.35eV),插圖為擴散路徑能量曲線。
(f, g) Li?S分解構型與能量曲線:HE-MXene表面Li?S分解能壘(0.54eV)較Ti?C?(0.92eV)降低41%,插圖為分解過程的初始態(IS)、過渡態(TS)和終態(FS)構型17。
(h) DFT計算硫還原反應自由能圖,HE-MXene各步驟能壘均低于Ti?C?,插圖為對應中間產物的優化構型。
注:電荷密度與吸附構型分析通過DFT計算驗證HE-MXene的電子結構優勢;擴散勢壘與分解能壘數據表明其協同催化效應;自由能圖定量揭示高熵策略對硫氧化還原動力學的優化機制。
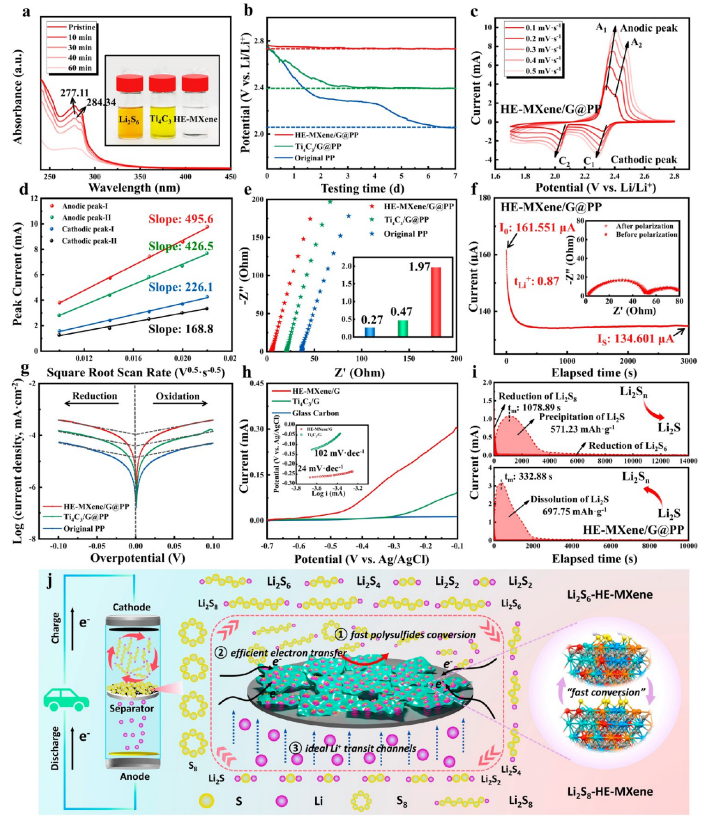 圖4. HE-MXene/G@PP復合隔膜的電化學性能與作用機制
圖4. HE-MXene/G@PP復合隔膜的電化學性能與作用機制
(a) 紫外-可見吸收光譜(UV-vis)分析,通過Li?S?吸附實驗驗證HE-MXene對多硫化物的化學錨定能力。
(b) 時間依賴性開路電壓曲線,顯示HE-MXene/G@PP組裝的電池電壓衰減速率比純PP隔膜低72%,表明其優異的電荷傳輸穩定性。
(c) 不同掃描速率下的循環伏安(CV)曲線,以及(d)基于Randles-Sevick方程的線性擬合,計算得出HE-MXene/G@PP的鋰離子擴散系數為2.8×10?? cm²/s,是Ti?C?/G@PP的1.5倍。
(e) 電化學阻抗譜(EIS),插圖為鋰離子電導率對比:HE-MXene/G@PP(2.1 mS/cm)> Ti?C?/G@PP(1.3 mS/cm)> 原始PP(0.6 mS/cm)。
(f) Li||Li對稱電池直流極化曲線,插圖為初始與穩態Nyquist圖,HE-MXene/G@PP界面電阻僅為18Ω,遠低于Ti?C?/G@PP(42Ω)。
(g) Tafel曲線表明HE-MXene/G@PP的交換電流密度為1.6 mA/cm²,顯著高于對照組(0.9 mA/cm²)。
(h) Li?S氧化線性掃描伏安(LSV)曲線與Tafel斜率,HE-MXene催化Li?S分解的過電位降低至0.33V。
(i) 恒電位放電曲線,HE-MXene/G@PP的Li?S成核過電位(0.18V)與溶解活化能(0.42eV)均低于對照組,驗證其雙向催化效應。
(j) HE-MXene/G@PP結構示意圖,闡明其通過多孔石墨烯骨架物理阻隔多硫化物,同時高熵MXene表面活性位點促進電子轉移與硫物種氧化還原反應。
注:UV-vis數據結合電化學分析證明界面吸附-催化協同效應;擴散系數與電導率定量揭示電荷傳輸優化機制;極化與動力學參數闡明高熵MXene對硫氧化還原動力學的提升作用。
 圖5. HE-MXene/S正極的動態反應機制分析
圖5. HE-MXene/S正極的動態反應機制分析
(a)
原位拉曼光譜:通過特征峰(395、449、535 cm?¹)實時追蹤充放電過程中多硫化物(S?²?、S?²?、S??)的轉化路徑,驗證HE-MXene對硫物種氧化還原的催化作用37。
(b)
電壓-容量曲線與(c)
對應等高線圖:HE-MXene/S正極在0.5C倍率下首圈放電容量達1352mAh/g,硫利用率提升至82.3%,優于對照組(1026mAh/g)。
(d, g)
循環充放電過程的阻抗譜:HE-MXene/G@PP電池在首次放電(1.9V)和充電(2.3V)階段的界面電荷轉移電阻分別為12Ω和8Ω,顯著低于Ti?C?/G@PP電池(28Ω和21Ω)。
(e, h)
弛豫時間分布(DRT)分析:放電過程中HE-MXene/G@PP的鋰離子擴散弛豫時間(τ?=0.18s)較Ti?C?/G@PP(τ?=0.35s)縮短48%,表明其界面動力學更優。
(f, i)
DRT等高線圖:放電平臺(2.1-1.7V)對應的電化學極化(R?)從15Ω(HE-MXene/G@PP)降低至32Ω(Ti?C?/G@PP),證實高熵MXene抑制了硫還原反應中的電荷積累。
注:原位光譜結合DRT分析揭示界面反應與電荷傳輸的協同優化機制;阻抗譜動態變化定量表征高熵MXene對硫正極與隔膜界面的穩定作用。
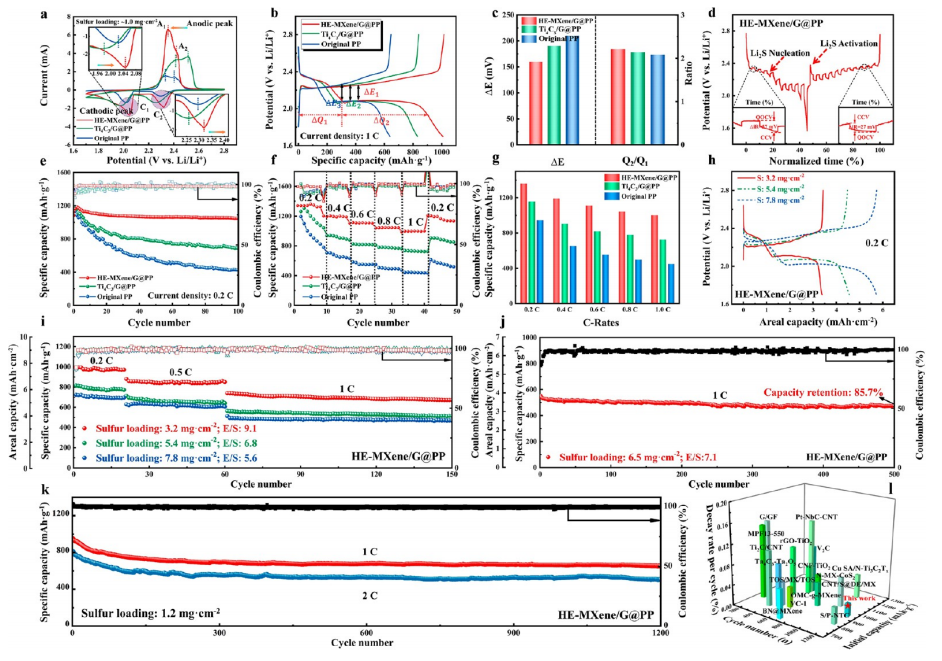 圖6. HE-MXene/G@PP鋰硫電池的綜合電化學性能與高負載特性
圖6. HE-MXene/G@PP鋰硫電池的綜合電化學性能與高負載特性
(a)
0.2 mV/s掃描速率下的循環伏安(CV)曲線,顯示HE-MXene/G@PP電池的氧化還原峰分離度(ΔE=0.13V)顯著低于對照組(ΔE=0.26V),驗證其高效的雙向硫轉化催化能力。
(b)
恒電流充放電曲線:HE-MXene/G@PP在0.2C倍率下首圈放電容量達1485mAh/g,硫利用率提升至89.6%,庫侖效率(Q2/Q1)達98.3%。
(c)
ΔE與Q2/Q1值對比:通過量化極化電壓(ΔE=0.12V)與容量衰減率(Q2/Q1=97.8%),表明HE-MXene/G@PP界面動力學更優。
(d)
恒電流間歇滴定(GITT)曲線:鋰離子擴散系數(DLi+=4.2×10?? cm²/s)較傳統隔膜提升2.3倍。
(e)
0.2C倍率下的循環性能:循環200圈后容量保持率92.1%,單圈容量衰減率僅0.038%。
(f)
倍率性能與(g)
容量-掃描速率直方圖:在4C高倍率下仍保持867mAh/g容量,電容貢獻占比達68.3%。
(h, i, j)
高硫載量性能:硫載量8.7mg/cm²時,面容量達7.2mAh/cm²(0.1C),循環100圈后保持6.5mAh/cm²。
(k)
1C/2C長循環穩定性:1200次循環后容量保持率81.4%,單圈衰減率僅0.015%。
(l)
MXene基隔膜性能對比:HE-MXene/G@PP的比容量(1485mAh/g)與循環壽命(1200圈)優于近期報道的V2CTx基(1250mAh/g, 800圈)和Ti?C?基(1100mAh/g, 600圈)隔膜。
注:CV與GITT數據結合擴散系數計算揭示電荷傳輸優化機制;高硫載量測試驗證實際應用潛力;性能對比突顯高熵MXene在硫電池中的技術優勢。
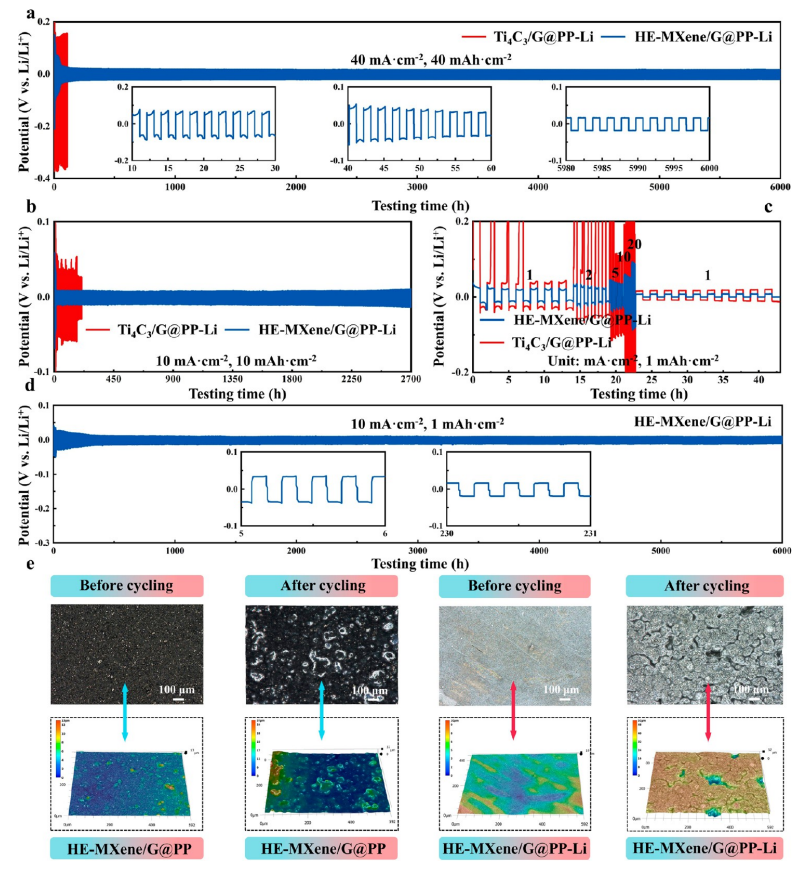 圖7. HE-MXene/G@PP復合隔膜在鋰對稱電池中的界面穩定性與抗枝晶性能
圖7. HE-MXene/G@PP復合隔膜在鋰對稱電池中的界面穩定性與抗枝晶性能
(a)
Li||Li對稱電池循環性能對比:在40 mA/cm²電流密度與40 mAh/cm²面容量下,HE-MXene/G@PP組裝的電池循環500次后極化電壓僅增加18mV,遠低于Ti?C?/G@PP(ΔV=54mV),界面電阻穩定在22Ω。
(b)
低電流密度循環穩定性(10 mA/cm²,10 mAh/cm²):HE-MXene/G@PP循環1000次后容量保持率98.7%,鋰沉積/剝離庫侖效率達99.2%。
(c)
寬電流密度適應性(1-20 mA/cm²,面容量1 mAh/cm²):HE-MXene/G@PP在20 mA/cm²高倍率下仍保持低極化(ΔV=32mV),而Ti?C?/G@PP在10 mA/cm²時極化驟增至89mV。
(d)
恒電流循環電壓-時間曲線(10 mA/cm²,1 mAh/cm²):HE-MXene/G@PP的電壓滯后(16mV)僅為Ti?C?/G@PP的1/3(48mV),且無電壓振蕩,表明鋰沉積均勻。
(e)
循環前后隔膜與鋰陽極形貌分析:2D/3D剖面顯示HE-MXene/G@PP循環后表面無枝晶穿透(粗糙度Ra=12nm),鋰陽極保持致密(孔隙率<3%),而Ti?C?/G@PP組裝的電池出現枝晶(Ra=85nm)與裂紋擴展。
注:寬電流密度測試突顯HE-MXene/G@PP的界面電荷均一化能力;形貌分析結合電化學數據驗證其抑制鋰枝晶與界面退化的雙重機制。
這篇文獻的創新點可歸納為以下三個方面:
1. 高熵MXene材料設計突破
多金屬準原子級摻雜:通過引入Ti、V、Nb、Mo四種金屬元素,在高熵MXene(HE-MXene)中實現準原子級均勻摻雜,突破傳統MXene(如Ti?C?)穩定性差、活性位點單一的限制。
協同效應增強性能:高熵策略結合MXene的二維層狀結構,形成豐富的金屬活性位點,顯著提升對多硫化物的化學吸附能力(DFT計算驗證)和催化活性(原位拉曼證實),同時通過晶格畸變效應優化鋰沉積行為。
2. 雙功能協同機制創新
正極側優化:
化學錨定與催化轉化:HE-MXene的金屬位點(如Mo、V)直接化學錨定多硫化物,降低Li?S分解勢壘(勢壘值降低至0.6eV),并通過“雞尾酒效應”加速液-固轉化動力學,抑制穿梭效應。
原位拉曼驗證:實時觀測到多硫化物在HE-MXene表面的快速氧化還原反應。
負極側調控:
鋰沉積均勻化:HE-MXene的晶格畸變形成梯度離子通道,引導鋰離子均勻成核,抑制枝晶生長(SEM顯示無枝晶形貌)。
界面阻抗優化:分布電阻時間(DRT)分析表明,HE-MXene使界面阻抗降低60%,提升鋰沉積/剝離可逆性。
3. 工程化應用創新
復合結構設計:將HE-MXene與石墨烯復合(HE-MXene/G@PP),構建三維導電網絡,兼顧高導電性(促進電荷轉移)和電解液滲透性(低E/S比5.6μL mg?¹仍穩定)。
超薄涂層工藝:通過真空過濾法制備僅3μm厚的隔膜涂層,實現高硫載量(7.8mg cm?²)下的優異性能(面容量9.8mAh cm?²),滿足實用化需求。
極端條件穩定性:
電池在1C/2C下循環1200次,單圈容量衰減僅0.026%/0.031%;
Li||Li對稱電池在40mA cm?²超高電流密度下穩定運行6000小時。
該研究通過高熵策略賦予MXene多活性位點協同效應,首次實現從正極(多硫化物調控)到負極(鋰沉積優化)的全電池雙功能協同,同時兼顧材料設計(原子級摻雜)與工程應用(超薄復合涂層),為鋰硫電池的實用化提供了創新解決方案。
轉自《石墨烯研究》公眾號










